|
|
||||||
|
|
|
|
Download | |||
Diamond Version 5 User Manual: Editing structural dataAtomic ParametersThe Atomic Parameters dialog has a report-like representation of atoms, which can be grouped for sites, with items that can be edited directly in the report
Read in this article:
Previous article: Symmetry and cell parameters About Atomic Parameters
The list of atomic parameters contains the positional as well as chemical informations about all atoms of the (crystal) structure.
For the regular case, that means, the compound is a crystal structure with translational symmetry,
the atoms in the parameter list describe only a set of atoms which is reproducible for the entire lattice by symmetry operations and cell translations.
In that case this set of atoms is often called the "asymmetric unit". Each atom in the "real world" of the crystal structure can thus be reproduced from an atom of the asymmetric unit by defining a code describing the symmetry operations to produce this atom. The coordinates of the atoms of the asymmetric unit refer to the base vectors of the unit cell and are thus fractional (x/a, y/b, z/c) and ideally (but not necessarily) in the range 0 <= {x,y,z} < 1. To add atoms to the parameter list, delete atoms, or to change parameters, choose the Atomic Parameters command from the Structure menu.
Diamond has no restrictions for the total number of atoms in the parameter list.
Parameters of each atom
For each atom, the following parameters can be defined:
Mandatory parameters
The following parameters of an atom are mandatory:
(a) If the atom site has valid coordinates (determined by diffraction or calculated), x, y, and z are mandatory. If the site is no dummy site (see below), a valid element symbol must also be given. Oxidation number, site occupation factor, number of attached hydrogens are not necessary to be given. The Wyckoff symbol will be calculated automatically by Diamond when the Atomic parameters dialog is closed with OK.
(b) If the position of the atom has not been determined, the coordinates x, y, and z are undefined. In most cases this "atom" describes H atoms which have not been localized in the unit cell. In such cases the site occupation may exceed 1. If e.g. the site occupation is 5 and the multiplicity is 4, there are 5 * 4 = 20 atoms in the unit cell with no positional parameters.
Dummy atoms
The element of the atom site (and thus the ordinal number) is defined by the element symbol, including 'D' and 'T' for deuterium and tritium, rsp. A dummy atom, i.e. an atom with well-defined site but without chemical meaning, may be defined by entering a '*' as element symbol (or leaving it blank). Dummy atoms represent a special kind of atom type. A dummy atom is useful to define coordination polyhedra with no central atom.
Mixed sites
If a compound contains mixed sites, you must specify an atom for each part of the site. Example: The site with coordinates (0; 1/2; 0.2345) is occupied 25 percent by Si and 75 percent by Al. In this case two atoms must be defined in the parameter list: Si with a site occupation factor of 0.25 and Al with a site occupation factor of 0.75. For more information, see the article "How mixed sites are determined and treated...".
Formula sum, calculated formula mass and density Diamond uses the atoms in the parameter list also to determine the formula sum, the formula mass and the density.
Changing position in parameter list
The order of the atoms in the parameter list can be changed by dragging the actually marked atom to the desired position.
Changes in the structure picture
If a structure picture has already been defined, changes in the atomic parameter list can have different effects on the picture, since all created atoms base upon the atoms of the parameter list:
Adding an atom to the parameter list:
This has no effect on the picture, since Diamond never creates automatically atoms from the parameter list.
Deleting an atom from the parameter list:
All created atoms basing upon the deleted atoms will be destroyed.
The position of an atom has been changed:
The coordinates of all atoms basing upon the changed atom will be recalculated. This may result in "strange distortions". Please note that bonds will not be affected. That means, no bonds will be destroyed, if the bonds get "too long" or "too short". Positions do not split if the multiplicity of an atom site increases, e.g. from a special to the general site. On the other hand, two or more atoms may share one and the same position, if the multiplicity decreases, e.g. from the general site to a special site.
The element has changed:
The elements of all corresponding created atoms change. But the designs must be updated manually according to the new atom type of the created atom. Note that on any changes in the atomic parameter list the lists of atom groups and bond groups will be rebuilt and thus connectivity will be reset!
Atomic Parameters dialogThe atomic parameters dialog shows the atoms as a report with the properties (label, Wyckoff position, coordinates) in several columns and items directly editable. Atoms can be arranged for sites (to group mixed site components). Symmetry-equivalent positions are recognized and specially color-coded. To open the atomic parameters dialog, use the Atomic Parameters command from the Structure main menu.
The following screenshot is taken from: COD:9013698:
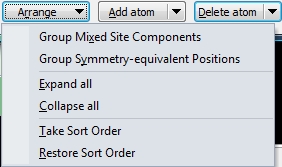
Arranging atoms or sites in the parameter list
The components of a mixed site can be grouped (as in the example above) or not. (The same is for symmetry-equivalent positions, but these are not available in COD:9013698.) The components can be expanded individually via the plus/minus signs or collectively using the Expand all or Collapse all command, rsp. You can change the sort order of the atoms in the parameter list by dragging and dropping a row. To make a new sort order permanent, run the Take Sort Order command. Use the Restore Sort Order to return to the original order of the atomic parameter list before the dialog was opened. Adding or deleting atoms or sites
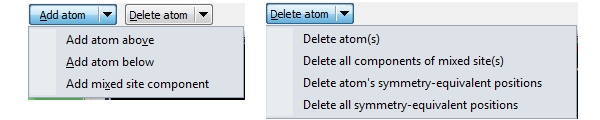
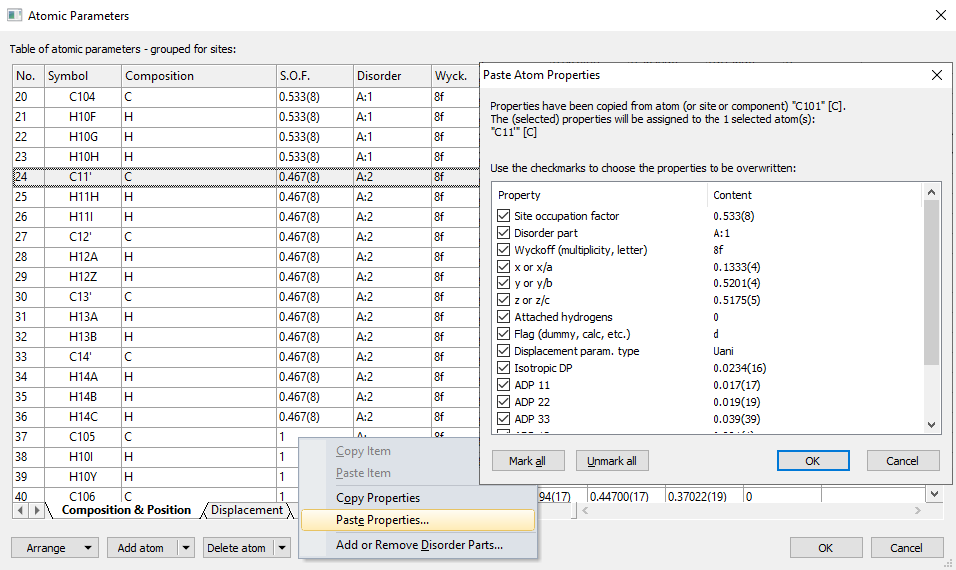
If you want to add an atom or a site to the current list of atoms (or sites), use the Add atom button directly or one of the commands from the Add atom drop-down menu (which opens when you click on the arrow-down symbol right beneath the Add atom button face). To delete the atoms currently marked in the table or some or all components or symmetry-equivalent positions, use Delete atom or the corresponding drop-down menu. Copying and pasting properties in Atomic Parameters dialogAn Edit button is available in the Atomic Parameters dialog offering several commands from a dropdown menu
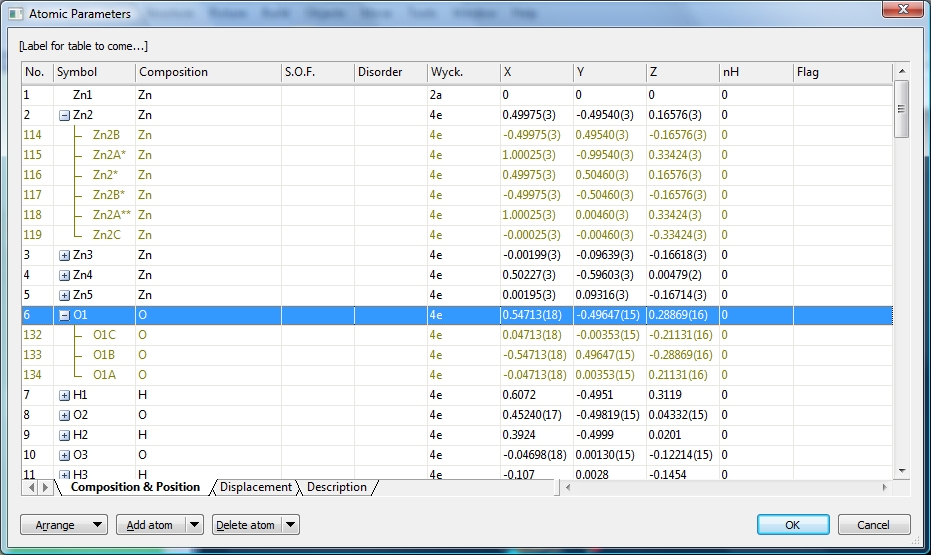
Symmetry-equivalent positionsIdeally, the atomic parameter list describes the asymmetric unit of the crystal structure, and symmetry-equivalent positions are created with suitable symmetry operations and/or integer cell translations. But the atomic parameter list may contain symmetry equivalent positions of the "asymmetric" atoms. These symmetry-equivalent positions often appear in CIF files imported from Cambridge Structural Database (CSD), in order to describe complete molecules when the asymmetric unit only reflects a fraction of those molecules. These positions, often labelled with additional letters and/or numbers and/or asterisks, are marked separately in an olive color and can be grouped together with their asymmetric atom.
The following screenshot is taken from a CIF file, exported from CSD (not available
with the Diamond sample files):
Since the most probable reason why symmetry-equivalent atoms are mentioned in the parameter list -- although redundantly -- is to easily create a complete molecule or a set of molecules, if wanted, when the asymmetric unit only contains an asymmetric part of one or more molecules in the cell. The command of choice to create the suggested set of atoms, is to run the command Add All Atoms and Connections from the Build menu. For details read the article "Adding all atoms (and bonds) of the parameter list(s)". Table of atomic parametersThe data pane, which is usually shown right beneath the structure picture pane, shows the data sheet by default. The atomic parameters are listed in both brief and comprehensive configuration. To have a table of atomic parameters instead of the data sheet, run the command View -> Table -> Table of Atomic Parameters. The table has the advantage that you can select one or more rows in the table, using the command Select Atoms from the table's context menu, e.g. in order to select all symmetry-equivalent atoms presently available in the structure picture. Cf. the article "Tables of objects like atoms, bonds, molecules etc." about using tables to select objects in the structure picture or vice versa. Additionally you can configure the table (columns to show or to be hidden), using the command Table Settings from the context menu. Note: The table of atomic parameters shows atoms, not sites. To have a list of atom sites (may be grouped for atom groups, and with their associated symmetry-equivalent atoms currently in the structure picture), use the "atom list", available via the View -> Atom List command. Cf. the article "Atom list: Hierarchical list of atom groups, atom sites, and created atoms of a structure picture".
Previous article: Symmetry and cell parameters |
|
Page last modified September 06, 2023. Copyright © 2023 Crystal Impact GbR. All rights reserved. Contact Webmaster |
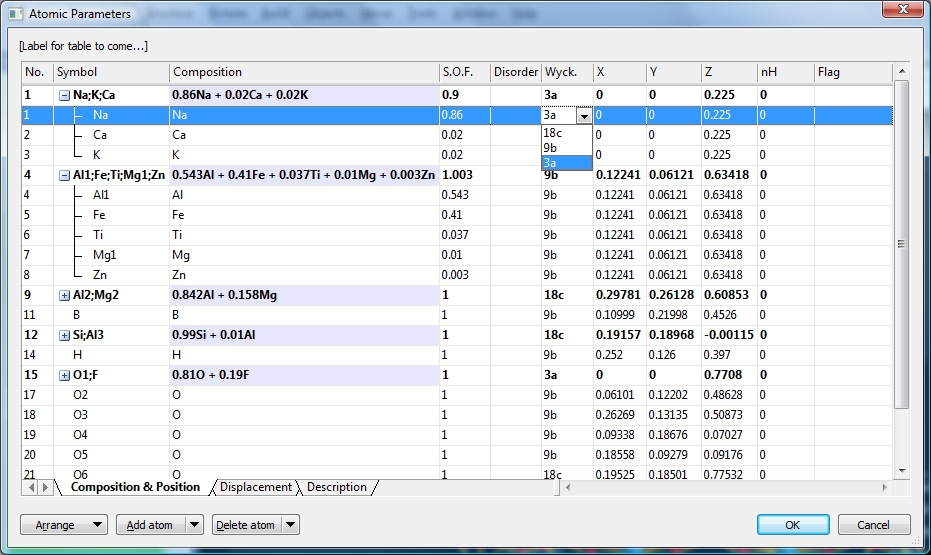 Screenshot of atomic parameters dialog of COD:9013698, oxy-schorl, formula sum:
Al6.741 B3 Ca0.02 F0.19 Fe1.23 H3 K0.02 Mg0.978 Na0.86 O30.81 Si5.94. The option
to group atoms on mixed sites is active (by default). Two of the mixed sites ("Na,K,Ca"
and "Al1,Fe,Ti,Mg1,Zn") have been expanded.
Screenshot of atomic parameters dialog of COD:9013698, oxy-schorl, formula sum:
Al6.741 B3 Ca0.02 F0.19 Fe1.23 H3 K0.02 Mg0.978 Na0.86 O30.81 Si5.94. The option
to group atoms on mixed sites is active (by default). Two of the mixed sites ("Na,K,Ca"
and "Al1,Fe,Ti,Mg1,Zn") have been expanded.