|
|
||||||
|
|
|
|
Download | |||
Diamond Version 5 User Manual: Editing structural dataSymmetry and cell parameters
Read in this article:
Previous article: Bibliographic data Space GroupThe symmetry of the crystal structure is defined by the space group. The space group is defined by the Hermann-Mauguin symbol plus an internal number to distinguish different settings of a space group. This number should not be mixed with the space group number defined in the International Tables for Crystallography, which has a range from 1 through 230.
A valid space group symbol is mandatory for a crystal structure in Diamond. Otherwise symmetry-induced atom positions cannot be calculated and therefore a crystal structure cannot be displayed correctly.
Diamond uses a space group information file that contains comprehensive informations for a total number of 670 space group settings. 530 of these space group settings are defined in Volume A (Space-Group Symmetry) of the International Tables for Crystallography. These 530 settings contain 230 standard settings plus additional settings for several monoclinic cell choices, unique axes, origin choices, or rhombohedral settings. Another set of 140 settings have been taken from the Inorganic Crystal Structure Database (ICSD), which are in parts identical with the 530 other settings but have special symbols to indicate e.g. the origin choice (e.g. "Fd3-mS", which corresponds to origin choice 1 in IT and is not centrosymmetric, whereas "Fd3-mZ" corresponds to origin choice 2 and is centrosymmetric).
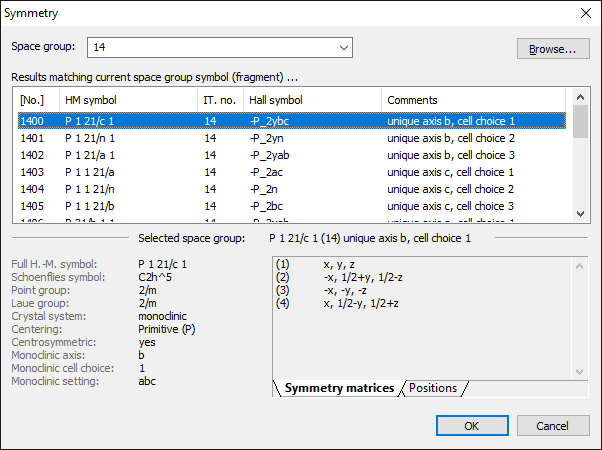
Entering a space group symbol or number To edit the space group, choose the Symmetry command from the Structure menu, which opens the Symmetry dialog.
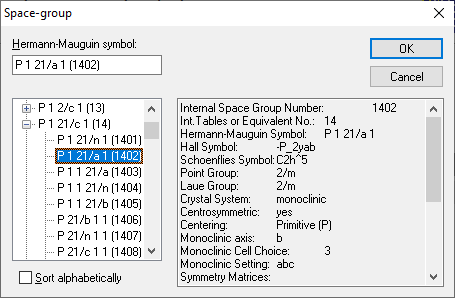
The screenshot shows a list of settings for International Tables space group number 14, with details about the marked setting in the lower half. The Symmetry dialog is to define the symmetry (space group) of the current structure parameter set by typing in a (Hermann-Mauguin) space group symbol or a space group number. Since there are multiple settings available each for many of the 230 space groups and there are different spellings, you can simply enter the space group number or at least a fraction of the Hermann-Mauguin space group symbol or - more general a crystal system. The space group(s) and/or settings matching the token in the input field Space group is/are listed in the Results table below the Space group input field. The table is updated automatically when you modify the token. For details about the search token as well as informations given for a (selected) space group, see the "online help" for the dialog, which is available via the F1 key. Browsing for a space group Instead of entering a space group symbol or number, you can use the Browse button to open the Space Group dialog where you can choose a space group from a hierarchical tree list of Hermann-Mauguin symbols and space group numbers that is sorted alphabetically or by International Tables space group numbers. The Space Group dialog is a versatile tool to select the right one out of the 670 space group settings. The 230 standard space groups are listed in a so-called tree view control. Additional settings are available by opening the corresponding node:
The example shows settings for space group no. 14. P 1 21/c 1 is the "standard" setting, whereas P 1 21/a 1 is highlighted. This setting has unique axis b and cell choice 3 like stated in the info field.
The Diamond space group info file contains the following informations for most of the space group settings. (Some informations such as special positions, Schoenflies symbol are not mentioned for the 140 space group settings from ICSD.)
This is an example for space group P 1 21/c 1 (no. 14):
"Only" the following informations about a space group setting are important for Diamond:
(1) The internal space group number. (2) The symmetry matrices of the general position in the right order (the sequence number of the symmetry matrix is used in the atom code).
(3) The Bravais translations according to centering, e.g. "+1/2,1/2,1/2" for I-centering.
(4) The symmetry matrices of the special position - together with multiplicities and Wyckoff letters - are used to calculate the correct Wyckoff positions in the atomic parameter list.
Other informations such as centrosymmetry, monoclinic cell choice etc. will not be used for the creation of atoms, but are helpful for the decision of the right setting.
Cell Parameters
The six cell parameters a, b, c, alpha, beta, and gamma define the dimensions of the unit cell. a, b, and c are the lengths of the unit cell base vectors, whereas alpha is the angle between the vectors b and c, beta the angle between a and c, and gamma the angle between a and b. a, b, and c must be given in Angstroem, the angles in degrees. For a crystal structure, which has translational symmetry, the cell parameters are mandatory, since they are necessary to calculate "real" coordinates from the fractional coordinates of the atomic parameter list, and thus to calculate distances and angles, thermal ellipsoids etc. correctly. To enter or change the cell parameters, choose the Cell Parameters command from the Structure menu. The cell parameters must fit with the crystal system of the actual (or previously set) space group. That will be no problem, if you begin with a new structure, since space group P1 is preset. But in most cases another space group has been defined, which may have restrictions to the cell parameters. The Cell parameters dialog has a special setting which disables all non-mandatory cell parameters. For example, b, c, alpha, beta, and gamma cannot be changed, if a cubic space group has been set. After closing the dialog with OK, Diamond checks if the six cell parameters fit the crystal system of the actual space group. For example, if you have entered a value for b which is different from a for a cubic cell (which is only possible, if the checkmark at Restrict cell parameters to crystal system has been cleared), you will be asked to change the space group. If a structure picture already exists, Diamond recalculates the coordinates of all created atoms. The connectivity will not be affected, but it is recommended to check the bond spheres, since the interatomic distances have changed due to the change of cell parameters.
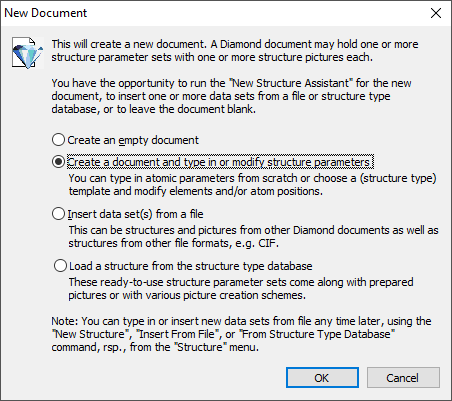
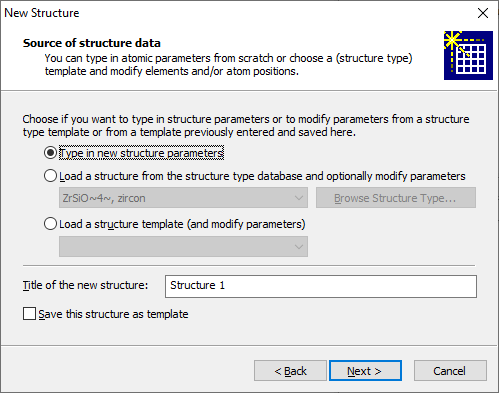
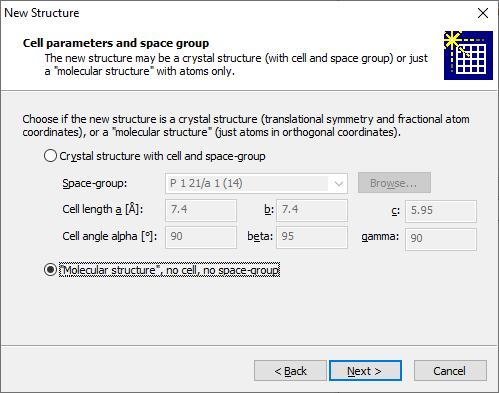
Structures with no cell and symmetry ("molecular structures")While the space group and cell parameters refer to a crystal structure and the "molecular structure" has neither cell parameters nor space group, you can define a "molecular structure" directly when you create a new Diamond document (or add another structure parameter set to an already existing Diamond document, since Diamond documents can hold multiple structure data sets with multiple pictures each, cf. the article "About documents, structures, and pictures"). Note: We use quotation marks for the "molecular structure" because it is not mandatory that the structure consists of (isolated) molecules. It can also be something amorphous, just the atomic coordinates are given in direct Angstroem coordinates and there is no cell to transform the coordinates and no space group to create atoms at symmetry-equivalent positions. To choose a "molecular structure" rather than the default crystal structure, choose the corresponding setting on the Cell parameters and space group page of the New Structure Assistant (third screenshot), which is followed by the New Document dialog (first screenshot) in response to the New command in the File main menu:
Note: To add a new structure to an already existing Diamond document, run the New Structure command from the Structure menu, which leads you directly to the New Structure Assistant.
Converting molecular structures into crystal structures and vice versaThs describes how to convert a "molecular structure", i.e. a structure data set with no translational symmetry (no cell parameters and no space group) into a crystal structure and the opposite way of removing translational symmetry (removing space group and cell information), i.e. to convert a crystal structure into a "molecular structure". There are two commands available in the Structure menu: The Add Translational Symmetry command creates a unit cell, transform the current orthogonal coordinates of the atoms to crystal coordinates, and applies space group symmetry. The origin of the crystal coordinate system will be placed in the origin of the current orthogonal coordinate system, with the a-axis parallel to the x-axis (pointing to the right). The b-axis will be placed in the xy-plane of the orthogonal coordinate system (i.e. in the plane of the screen). If the angle gamma is 90 degrees, the b-axis will be parallel to the y-axis. The c-axis will point towards you; if both angles alpha and beta are 90 degrees, the c-axis will be parallel to the z-axis of the orthogonal coordinate system. You enter the space group as well as the six cell parameters a, b, c, α, β, and γ in the Add Translational Symmetry dialog. Depending on the crystal system of the space group, some of the unit cell parameter input fields below will be fixed/deactivated. If there are overlapping/identical atoms after application of the space group symmetrs (i.e. if a certain groups of atoms can now be described by a single atom in the parameter list), the corresponding surplus atoms will be deleted from the atomic paramter list. The Remove Translational Symmetry command removes cell and space-group information from the current structure parameter set and converts atom coordinates from crystal or fractional (x/a, y/b, z/c) to orthogonal coordinates in Angstroem units.
Previous article: Bibliographic data |
|
Page last modified September 05, 2023. Copyright © 2023 Crystal Impact GbR. All rights reserved. Contact Webmaster |