Diamond 4: Extended functionality for molecules and polymers
< Previous: Enhanced productivity
Go to Diamond 4 Features Overview
Next: New polyhedron functions >
Several options to create packings of molecules
In addition to the "Complete fragments" function (already known in Diamond 3) that
lets you complete the molecular fragments that had been truncated, for instance
during a cell range filling, you can now define packings of molecules
much more straight-forward. Define a unit cell or a super cell or any arbitrary parallelepiped
of your crystal structure, or a sphere or a slab (a cell range cut by an hkl plane)
or a slice parallel to an hkl plane of given thickness as basis for the atoms and
use several options which atoms of the molecules are to be included in the packing range: only atoms that fit (which may cut off atoms outside) - or the whole molecule
if all or at least one atom fits
or if the center or a reference position of the molecules fits.
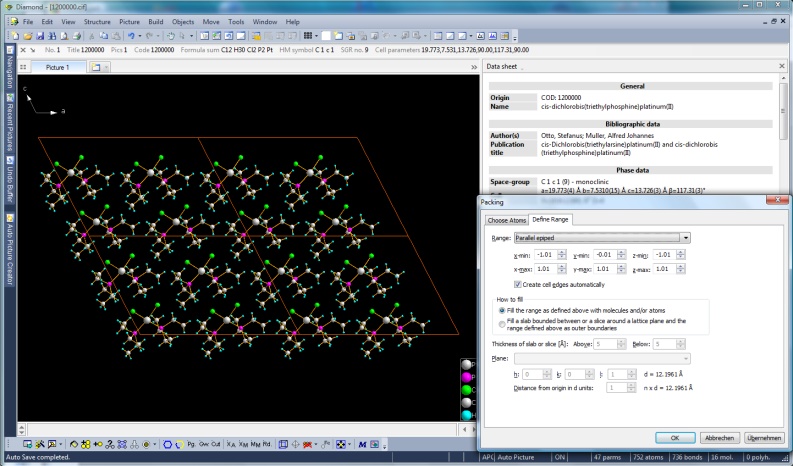
Packing diagram of cis-dichlorobis(triethylphosphine)platinum(II), COD: 1200000
(Full size image: 336 KB)
Non-bonding contacts and improved handling of H-bonds
The well-known "Connectivity" dialog where you define, if and how far atom group
pairs are to be connected with bonds, has got two additional pages, one for
non-bonding contacts and another one for H-bonds. The basic settings are derived
from the van der Waals radii sums but can be adjusted individually for atom group
pairs or even atom site pairs individually.
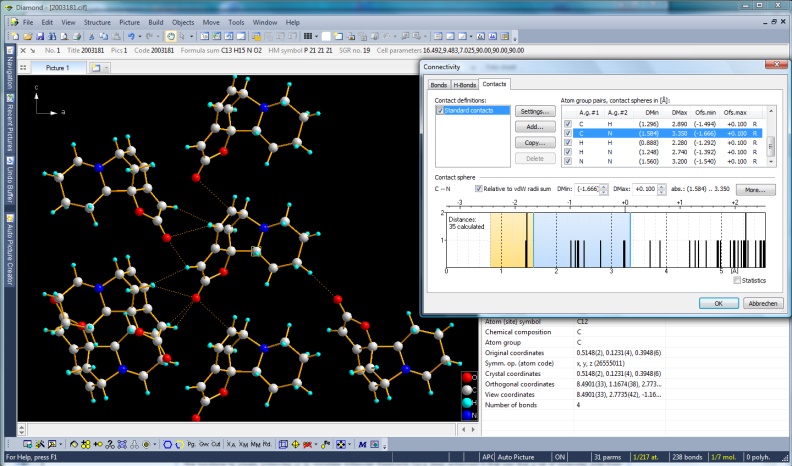
Cluster of molecules connected with non-bonding contacts. In the Connectivity dialog,
a dmax of van der Waals radii sum plus 0.1 A is set. Securinine, COD:2003181
(Full size image: 332 KB)
Connection parameters
Like atomic parameters, bond parameters can be defined now with the structure parameters.
These "hard" connections between (symmetry-equivalent) atoms of the parameter list
eventually supersede the settings made under "Connectivity" (the latter called "soft"
connections). They can be input manually but are normally imported via the "_geom_bond_xxx"
loop in a CIF file or via the bond section in a molecular structure file (e.g. a
MOL file). The same is for non-bonding contacts ("_geom_contact_xxx") and H-bonds
("_geom_hbond_xxx"). There are several options, if and how to consider the "hard"
connections in your connectivity settings.
Improved functions to complete molecular fragments and to create molecules
The functions to create molecules or to complete molecular fragments have been enhanced
in that way that a list of molecular units from the atomic parameter is available
to select from. This list is based upon the current settings of either connectivity (a bonding sphere for an atom group
pair each) or atomic environments. You can define
a reference atom within the molecular unit and choose symmetry operations and/or
cell translations to be applied to the molecular unit to create your symmetry-equivalent
molecules whereever you want to have it in your crystal structure picture.
Expand or reduce clusters of molecules
Expand or reduce clusters of molecules by finding neighbouring molecules (usually
via connection with non-bonding contacts and/or H-bonds - but also possible with
a simple "who's next?" search). You can create these neighbouring
molecules directly, or build them up step-by-step from the contacting atoms, optionally
using broken-off bonds.
Grow or cut molecular fragments or polymers
Especially for larger molecules or polymers, there is now the possibility to grow
or cut molecular fragments or polymers step-by-step - as a kind of missing link
between the Diamond 3 functions "Coordination spheres with N cycles" and "Complete
fragments".
Blow up or shrink multiple coordination spheres in polymers
Especially useful for polymers are the new functions to blow up or shrink multiple coordination spheres around selected atoms.
The mentioned functions can be used through commands from the "Build" menu but also
very intuitively using the mouse wheel button and the mouse wheel to expand or reduce
molecular clusters, grow or cut polymers, or blow up or shrink coordination spheres
around the atom(s) under the mouse cursor.
< Previous: Enhanced productivity
Go to Diamond 4 Features Overview
Next: New polyhedron functions >
|

