|
|
||||||
|
|
|
|
Download | |||
Diamond Version 5 User Manual: Building up structural partsSupport for disorder partsFunctions that connect atoms and search for neighbouring atoms or complete molecules consider, if and what disorder part the atoms belong to and avoid connections between atoms of different parts. The disorder informations usually come from "_atom_site_disorder_assembly" and "_atom_site_disorder_group" items in a CIF file and are formatted in Diamond with a ':' between, e.g. an atom from disorder assembly "A" and disorder group "1" has a disorder part "A:1". The "Filter" dialog offers a list showing all disorder parts of the current structure, making it easier to disable atoms from unwanted disorder parts to be considered in building operations like "Fille unit cell" or "Create molecules". The command "Select Disorder Part" ("Edit" menu) selects all atoms (and bonds between) belonging to the same disorder part as the selected atom. This is useful to edit atom and bond designs of disorder parts to differentiate them from other parts. Disorder information can easily be edited in the "Atomic Parameters" dialog and is shown in the info tip (mouse over atom) as well as in several dialogs (tables to select atom sites from) and as additional columns in table of created atoms and table of created bonds.
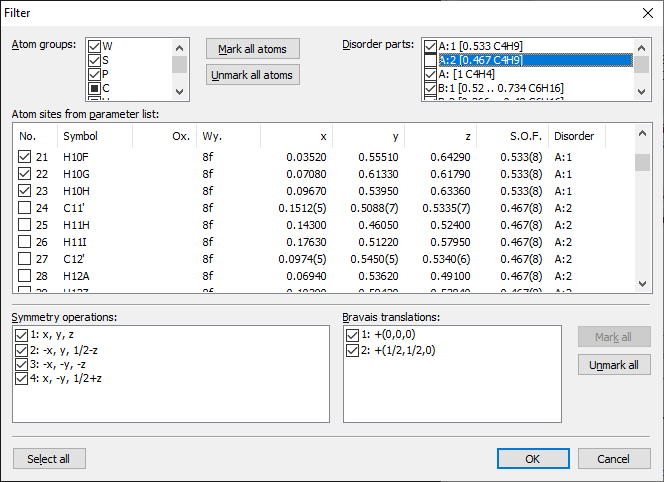
Previous article: Determination of atom site environments basing upon Dirichlet domains (Voronoi polyhedra) Using the Filter dialog to exclude atoms of unwanted disorder partsThe "Filter" dialog ("Build" menu) offers a list of all disorder parts of the structure. Using the checkmarks to "switch off" one or more disorder parts makes it easier to disable atoms from unwanted disorder parts to be considered in building operations like "Build/Fill/Fill unit cell" or "Build/Molecules/Get molecules". On the other side, functions that complete spheres or molecular fragments or grow polymeres automatically consider disorder part informations without the necessity to use a filter. The screenshot [1] shows the "Filter" dialog with the disorder part "A:2" disabled. The atom site list below reflects the current de-selection of the constituent atoms.
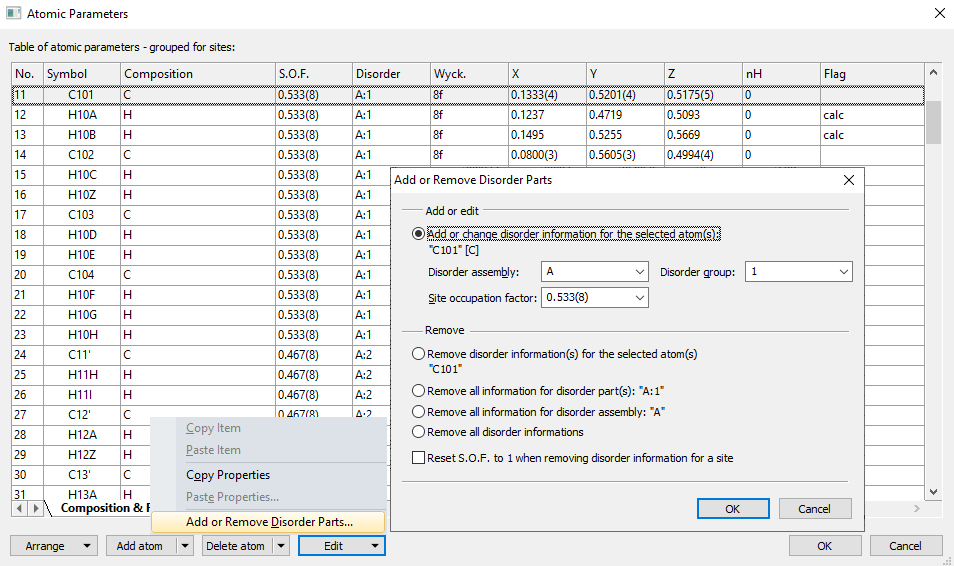
Selecting disorder parts and editing their atom and bond designsThere is a command "Select Disorder Part" in the "Edit" menu, which selects all atoms (and bonds between) that belong to the same disorder part as the atom or atoms that are currently marked as selected in the structure picture. The main purpose to select a disorder part's atoms and bonds is to edit their designs to differentiate between this disorder part and other disorder parts or the non-disordered rest of the structure. To edit the atom and bond designs after selecting a disorder part, at best run the command "Edit/Atom Design..." or "Edit/Bond Design..." from the structure picture context menu. You may use the new command "Set opacity to S.O.F." in the "Atom Design" dialog and the corresponding option (checkbox) "Adjust bond transparency to atoms' transparencies" in the "Bond Design" dialog. Besides this, you can change any other properties, e.g. line styles to dotted - especially when you are creating a flat mode picture. While the "Select Disorder Part" command selects only the bonds where both atoms belong to the same disorder part, you can extend this selection with the "Extend Selection" command, which then selects the bonds from disordered atoms to the non-disordered neighbouring atoms, too. Adding (or removing) and showing disorder informationsThe "Atomic Parameters" dialog ("Structure" menu) has an "Edit" dropdown menu button with several commands - amongst them commands to copy and paste items or whole atom properties - and an "Add or Remove Disorder Parts" command for easier adding, editing or removing of disorder part information to or from the atomic parameter list. The "Add or Remove Disorder Parts" dialog offers the option to add disorder information to the atoms that have been marked as selected in the atomic parameters table. You can choose from disorder assemblies and groups as well as site occupation factors that have been defined up to then for the whole atomic parameter list or define new disorder parts. On the other side, you can remove disorder information for selected atoms or for the disorder parts or assemblies where at least one atom is selected from - or all disorder information.
Disorder information is shown:
Regarding disorder parts in distances calculation
Previous article: Determination of atom site environments basing upon Dirichlet domains (Voronoi polyhedra) [1] COD: 1004001. Jin, Song.; Zhou, Ran.; Scheuer, Ellen. M.; Adamchuk, Jennifer.; Rayburn, Lori. L.; DiSalvo, Francis. J.; "Synthesis, Characterization, and Ligand Exchange Studies of W6S8L6 Cluster Compounds"; Inorganic Chemistry, 40, 2666-2674 (2001) |
|
Page last modified July 7, 2022. Copyright © 2022 Crystal Impact GbR. All rights reserved. Contact Webmaster |