Atomic Environments
"Atomic environment" as additional (optional) criterion when
filling a coordination sphere or adding coordination polyhedra
Atomic environments in brief:
- Definition of atomic environments for an asymmetric atom site each enables more
precise definition of neighbouring atoms connected with strong bonds than just the
connectivity which is defined between atom groups and by rather simple two sphere
boundary values rmin and rmax each.
- Dialog "Build/Atomic Environments" to edit (add or remove) atoms from the central
atom's environment each. Two presentations: Distances table and histogram.
- Correlation (optional, by default: yes) between atomic environments and connectivity
(bonding spheres from atom group pairs).
Previous article: Connectivity, part 1: Bonding spheres
Next article: Determination of atom site environments basing upon Dirichlet domains (Voronoi polyhedra)
Why atomic environments?
Defining an atomic environment for every atom site makes the search for bonded neighbouring
atoms much more flexible than by defining between atom types only. This is especially
the case when some of the atom sites belonging to a certain atom type have deviating
environments, and a bonding sphere definition in the "Connectivity" dialog does
not match all cases. Or the spreading of distances in the histogram of the "Connectivity"
dialog makes it difficult to find the (upper) boundary of the bonding sphere for
the selected atom type pair there.
As an example, the distances histogram for our sample "PCD-1251073.diamdoc", which is a 1:1
adduct of SbF3 and SbF5, the distances distribution around the Sb+3 atoms arises
the question: There is a gap, but where ends the bonding sphere really?
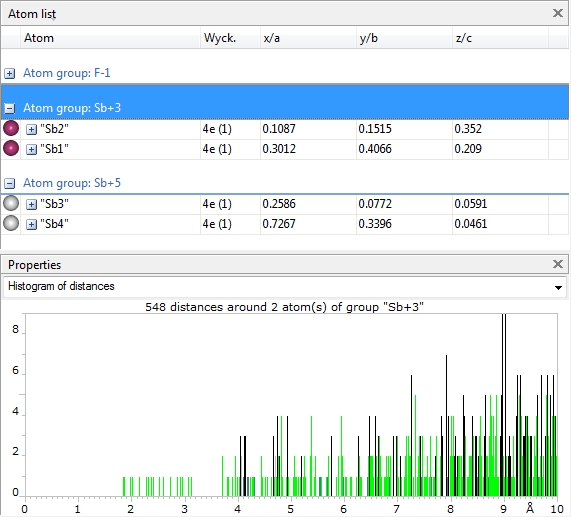
Screenshot of the atom list for PCD-1251073.diamdoc showing a distances histogram around atoms belonging to atom type Sb+3
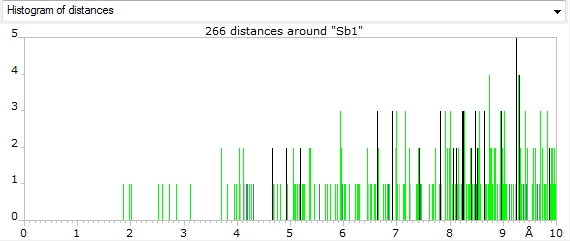
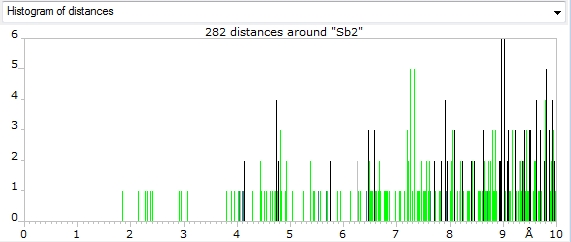
Screenshots of distances histograms for atom sites "Sb1" and "Sb2", which are associated
with atom type Sb+3
"Atomic Environments" dialog
You find the new "Atomic Environments" dialog next to the "Connectivity" command
in the "Build" menu. to edit (add or remove) atoms from the central atom's environment
each. Two presentations: Distances table and histogram.
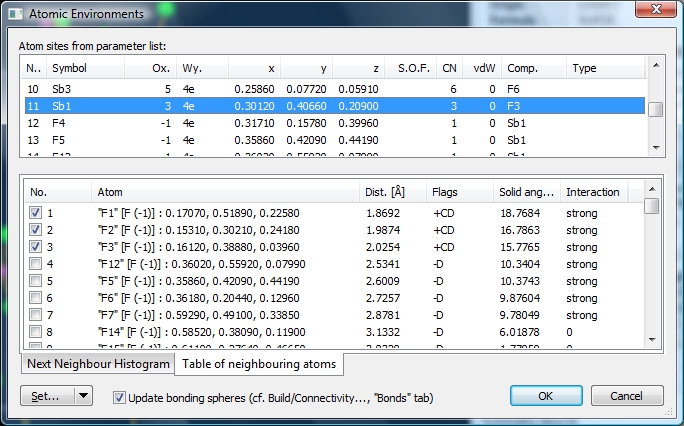
The upper half of the dialog shows the list of all atom sites of the current structure,
whereas the lower half lists atoms in the neighbourhood to the selected atom site
up to a distance of 6 Angstroms. The neighbouring atoms are the atoms belonging
to the Dirichlet domain of the selected atom site, followed by the residual atoms
up to the mentioned 6 Angstrom boundary. A checkmark in the first column indicates
if the neighbouring atom belongs to the atomic environment or not.
The "Flags" column uses the following signs:
- A plus or minus sign indicates that the atom belongs to the atomic environment
and corresponds with the checkmark, unless you manually set or clear a checkmark
to manually add or remove an atom to or from the atomic environment.
- 'C' means that the
neighbouring atom matches the bonding sphere of the corresponding atom type pair
(for instance Sb+3--F-1) in the connectivity (cf. "Build"/"Connectivity...").
- 'D' means that the neighbouring atom belongs to the Dirichlet domain (which does
not necessarily mean there is a strong interaction with the central atom, since
there are additional conditions for that).
- 'P' means that the neighbouring atom is defined as bonded to the central atom
in the bond parameter list, that means in the structural parameters. Cf. "Structure"/"Connection
Parameters..." and the article "Connection Parameters".
"Solid angle" and "Interaction" deal directly with Dirichlet domains. Read the article
"Dirichlet Domains" for details.
Setting or resetting atomic environments
There are two commands available through
the dropdown arrow right of the "Set..." push button to set or
reset all atomic environments:
- "Reset to Connectivity Settings" takes all atoms matching the
bonding spheres that are defined on the "Bonds" page of the "Connectivity" dialog
and removes the atoms outside the bonding spheres or if the associated atom group
pair is not defined as bonding anyway.
- "Set Dirichlet Domains" considers Dirichlet domains as well as
the overlap of deriving "spherical domains" and "Slater radii". Cf. the articles
"Dirichlet Domains" and
"Voronoi Polyhedra" for details.
Correlation between bonding spheres and atomic environments
By default, changes in the atomic environment definitions update the sizes of the
bonding spheres, which are defined for pairs of atom groups, and can be checked
and edited on the "Bonds" page of the "Connectivity" dialog (command "Build"/"Connecitivity...").
And vice versa, changes of bonding spheres update the atomic environments. Since
atomic environments (on atom site level) are more exact than bonding spheres (on
atom type/group pair level), it can happen that atomic environments and connectivity
lead to deviating results.
You can clear the "Update bonding spheres[...]" checkmark in the "Atomic Environments"
dialog and/or the corresponding checkmark in the "Connectivity" dialog to advertently
define atomic environments different from bonding spheres for special reasons, for
instance to define special coordination polyhedra -- and using "Atomic Environment"
sphere type in "Add Polyhedra" dialog whereas using "Generic" sphere type in the
other dialogs ("Connect atoms", "Define Molecules", "Coordination Spheres"). In
any case, you should know what and why you do.
Previous article: Connectivity, part 1: Bonding spheres
Next article: Determination of atom site environments basing upon Dirichlet domains (Voronoi polyhedra)
Reference for PCD-1251073:
Sb4F16 in P121/c1 (14);
"The Crystal Structure of the 1:1 Adduct of Antimony Trifluoride and Antimony Pentafluoride",
by: Gillespie R.J., Slim D.R., Vekris J.E.; J. Chem. Soc. (Dalton Trans.) 1977,
p. 971.
|
