
|
||||||
|
|
|
|
Download | |||
Diamond Developer BlogUsing bond, H-bond, and contact parametersKlaus Brandenburg -- 9 May 2013, 22:00 UTC This article demonstrates several direct usages of connection parameters, i.e. usually basing upon _geom_bond_xxx, _geom_contact_xxx and _geom_hbond_xxx informations imported from a CIF file. We use the compound COD:1501635 (Ulrich Darbost, Janie Cabana, Eric Demers, Thierry Maris, James D. Wuest; CheM, 1 (2011), 52-12369) for demonstration.
You have been knowing the table of atomic parameters in the data pane right beneath
the structure picture view since Diamond version 2. This table is now supplemented
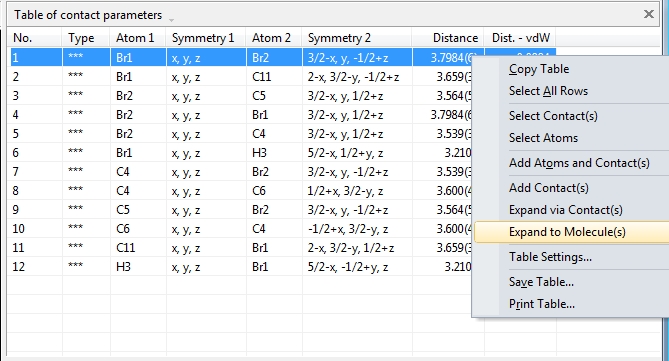
by three more tables: Like the atomic parameters, these connection parameters can be edited using the command "Connection Parameters..." from the "Structure" menu. The dialog has three tabs (pages) for the bonding, contacting and H-bond parameters, rsp. The tables are available through the "View"/"Table" sub-menu or from the corresponding dropdown menu in the right part of the main toolbar. The screenshot shows COD:1501635 with table of contact parameters and "Connection Parameters" dialog open with table of bond parameters: 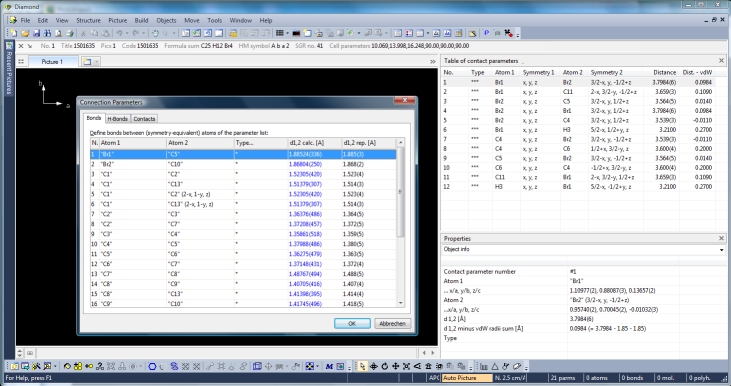
In some cases, the bond parameters are used to strike out several bonding spheres, spheres that may differ from those generated by effective radii, for instance. But in many other cases, the bond parameters, together with the atomic parameters, can serve as a simple starting point to create molecule(s) directly from the atomic parameter list. This is done in Diamond with the command "Add All Atoms" from the "Build" menu. This also considers non-bonding contacts and H-bonds when mentioned in the structure input parameters. The following picture of COD:1501635 is the result of command "Build"/"Add All Atoms" (which in this case also involves the contact parameters). The bond parameters (and contact parameters) consider only a part of the spirobifluorene molecule. This is a case where the bond parameters do not automatically create complete molecule(s). (Here we have 25 bonds and 12 contacts. It is somewhat more than the "asymmetric" half of the spiro molecule.) 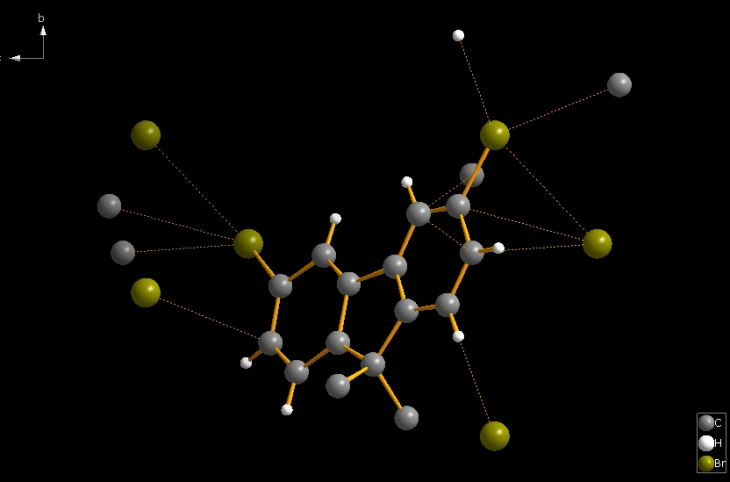
So -- as in general in Diamond for molecular structures -- the command "Create Molecules Directly" (or Shift+Ctrl+M) serves better. The second picture shows the result of "Create Molecules Directly" plus "Create Atoms' Neighbouring Atoms" (Shift+Ctrl+X; the "expand" command from the toolbar, cf. Related Articles). Then the result has 46 bonds and 24 contacts. 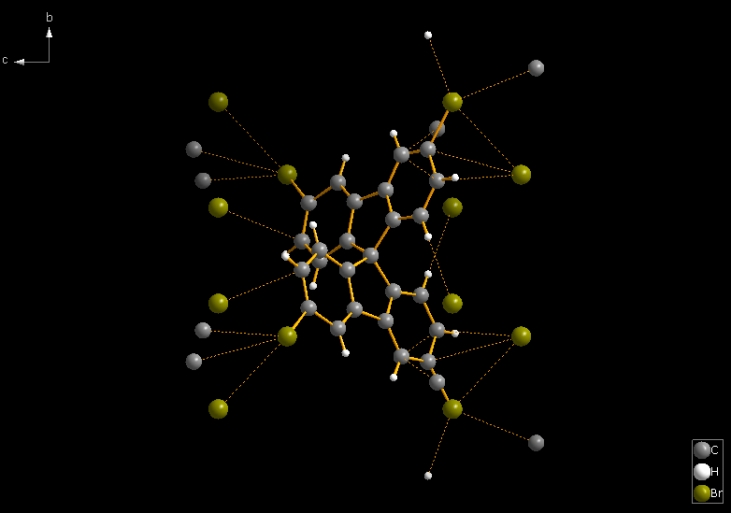
The following shows how to expand from a molecule (made by "Create molecule directly" or Shift+Ctrl+M) via one contact definition from the table of contact parameters, here the first mentioned: Br1 -- Br2. This leads to the final picture with five molecules. 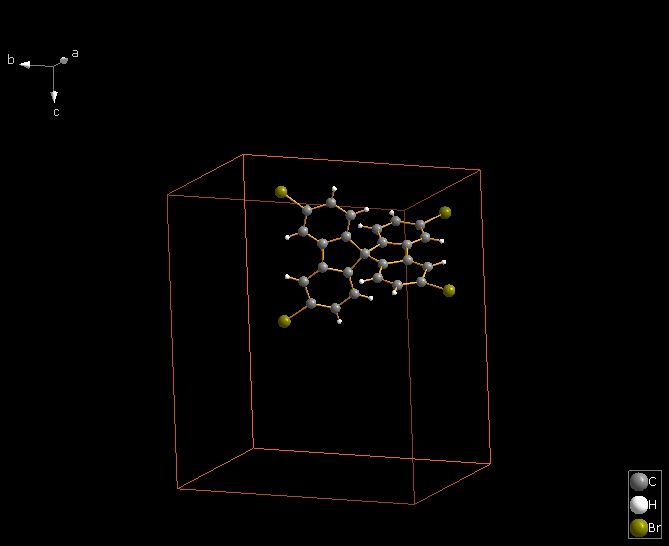
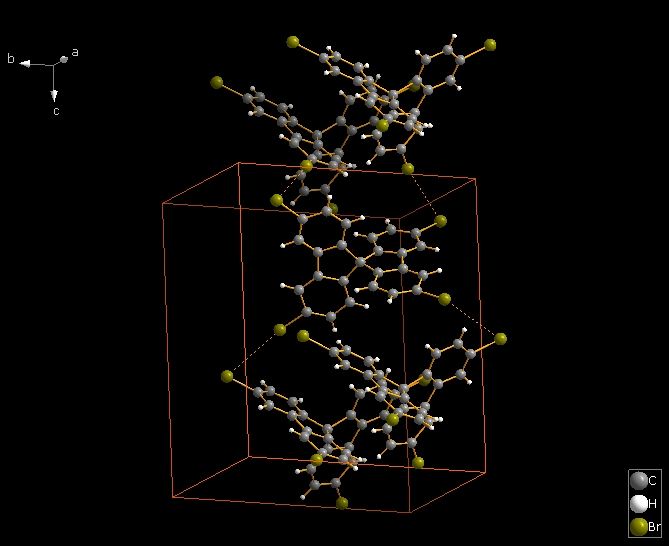
Just to mention: H-bond parameters serve in the same way, but here in COD:1501635 there is just one H-bond definition. |
|
| Page last modified May 15, 2018.
Copyright © 1997-2018 Crystal Impact GbR. All rights reserved. Imprint / Contact Privacy policy Webmaster Google+ Twitter |